Henning Schulzrinne, the Julian Clarence Levi Professor of Mathematical Methods and Computer Science, has been named a 2014 ACM (Association for Computing Machinery) Fellow in recognition of “contributions to the design of protocols, applications, and algorithms for Internet multimedia.” He is one of 47 to be so recognized in 2014 for contributions to computing.
“I’m deeply honored to be named ACM Fellow. Indirectly, this also recognizes the contributions of all the IRT lab members over the years,” says Schulzrinne, in referring to the Internet Real-Time Lab (IRT) lab, which he directs and which conducts research in the areas of Internet and multimedia services.
It is the second honor accorded Schulzrinne in as many months. In December he received an Outstanding Service Award by the Internet Technical Committee (ITC), a joint committee of the Internet Society and the IEEE Communications Society. The award was given in recognition of excellent and extended service rendered to the fields of the ITC, of which Schulzrinne was the founding chair. He served from 1994 to 2000 and helped shape the agenda of the committee in its early years.
The ITC award, being given for the first time, acknowledges both Schulzrinne’s service to the ITC as well as service to the community through his foundational work on the key protocols Session Initiation Protocol (SIP) and Real-Time Transport Protocol (RTP) that led to the development of Voice-over-IP (VoIP).
The two awards are testament to his lasting achievements and the immense impact of VoIP and related technologies on society, and they follow on Schulzrinne’s induction into the Internet Hall of Fame in 2013. Previous awards include the New York City Mayor’s Award for Excellence in Science and Technology and the VON Pioneer Award. He is also an IEEE Fellow.
In addition to his research, Schulzrinne is active in public policy and in serving the broader technology community. From 2011 until 2014, he was the Chief Technology Officer for the Federal Communications Committee where he guided the FCC’s work on technology and engineering issues. He continues to serve as a technical advisor to the FCC. Schulzrinne is also a past member of the Board of Governors of the IEEE Communications Society and a current vice chair of ACM SIGCOMM. He has served on the editorial board of several key publications, chaired important conferences, and published more than 250 journal and conference papers and more than 70 Internet Requests for Comment.
Currently a professor within Columbia’s Computer Science department, Schulzrinne continues to work on VoIP and other multimedia applications. Ongoing projects look to create an overall architecture for the Internet of Things, limit phone spam (“robocalls”), make it easier to diagnose network problems, and contribute to a better understanding between engineers and public policy.
He is widely known for developing key protocols, including the Session Initiation Protocol (SIP), that enable Voice-over-IP (VoIP) and other multimedia applications that are now Internet standards. His research interests include Internet multimedia systems, applied network engineering, wireless networks, security, quality of service, and performance evaluation.
Before joining the Computer Science and EE departments at Columbia University, Schulzrinne worked at AT&T Bell Labs and was associate department head at GMD-Fokus (Berlin).
Education
Undergraduate degrees in economics and electrical engineering, Darmstadt University of Technology, Germany
MSEE degree as a Fulbright scholar, University of Cincinnati, Ohio PhD, University of Massachusetts, Amherst
Awards and recognitions
2013, Internet Hall of Fame
2010, 2011, IEEE William Terry Lifetime Distinguished Service Award
2006, IEEE Fellow
2004, Mayor’s Award for Excellence in Science and Technology (NYC)
2000, NSF Young Investigator Award, VON Pioneer Award
Bjarne Stroustrup, a visiting professor at Columbia Engineering, is the senior award winner of the 2015 AITO (Internationale pour les Technologies Objets) Dahl-Nygaard Prize for the design, implementation, and evolution of the C++ programming language. For this same achievement, Stroustrup has also been made a Fellow of the Computer History Museum.
This prize, established in 2005 and one of the most prestigious in the area of software engineering, is named for Ole-Johan Dahl and Kristen Nygaard for their work in creating Simula, the first object-orientated language.
“I feel particularly honored by this award because I knew Ole-Johan Dahl and Kristen Nygaard,” says Stroustrup. “While still a student in the University of Aarhus, I learned object-oriented programming from Kristen Nygaard through long discussions.”
Stroustrup was influenced by the object-oriented model of Simula when he first began work on C++ in 1979. Then at Bell Labs, he needed a language that would provide hardware access and high performance for systems programming tasks while also handling complexity. Since no such language then existed, he designed one by essentially building on top of C to add support for object-oriented programming, data abstraction, and other capabilities.
In 1985, C++ was commercially released and spread rapidly, becoming the dominant object-oriented programming language in the 1990s and one of the most popular languages ever invented.
Fittingly Stroustrup is the recipient of many awards, honorary degrees, and other recognitions. He is a member of the National Academy of Engineering and was previously made a Fellow of both the IEEE and the ACM. Just recently, he was named also a Fellow of the Computer History Museum to recognize the impact and importance of C++ in computer history.
Stroustrup continues to update and add functionality to C++. Even as new languages have been created for the rapidly shifting programming landscape, C++ remains widely used, particularly in large-scale and infrastructure applications such telecommunications, banking, and embedded systems.
About Bjarne Stroustrup
Bjarne Stroustrup is the designer and original implementer of C++, the widely used programming language that has significantly impacted computing practices and pushed object-oriented technology into the mainstream of computing.
In addition to his continuing work on expanding C++, Stroustrup is interested in researching distributed systems, design, programming techniques and languages, and software development tools.
Now a Visiting Professor in Computer Science at Columbia, Stroustrup is also a Managing Director in the technology division of Morgan Stanley, and continues to serve a Distinguished Research Professor in Computer Science at Texas A&M University as he has since 2002.
Education
Master’s degree in mathematics and computer science (1975),Aarhus University, Denmark.
PhD in computer science (1979),
University of Cambridge.
Awards and recognitions (a sampling)
2013
Electronic Design Hall of Fame. Honorary Doctor of Computer Science from The National Research University, ITMO, St. Petersburg, Russia.
The Golden Abacus Award from Upsilon Pi Epsilon.
2010-2015
Honorary Professor in Object Oriented Programming Languages, Department of Computer Science, University of Aarhus.
2010
The University of Aarhus’s Rigmor og Carl Holst-Knudsens Videnskapspris. The university’s oldest and most prestigious honor for contributions to science by a person associated with the university.
2008
Dr. Dobb’s Excellence in Programming award.
2005
The William Procter Prize for Scientific Achievement from Sigma Xi (the scientific research society).
2005
IEEE Fellow.
2004
Elected member of The National Academy of Engineering.The IEEE Computer Society’s Computer Entrepreneur Award.
2002-2006
Honorary Professor at Xi’an Jiao Tong University.
1996
AT&T Fellow.
1995
Named one of “the 20 most influential people in the computer industry in the last 20 years” by BYTE magazine.
Identifying the specific genes that cause a cancer—the so-called driver genes—is a central goal in cancer research. The more drivers identified, the more options to treat the cancer with a drug known to act on that gene. But identifying a specific driver is not easy, especially when entire sections of the chromosome are damaged. Traditional biological tools, slow and imprecise, have identified only a limited number. As cancer “big data” accumulates, researchers are turning to computational methods, specifically machine learning, to identify additional drivers. But it’s not immediately clear how to apply these methods to genetic data. Machine learning expects a lot of data, but biological data is relatively sparse compared to other application domains in machine learning. In a paper published last November in Cell, Felix Sanchez-Garcia, a computer science PhD student, describes a new algorithm that integrates data from multiple diverse sources, using the strong features in one to bolster weaker evidence in the other. Applied to breast cancer patients, the algorithm doubled the number of identified driver genes.
Cancer is a heterogeneous disease, and no two cancers are the same even when they occur at the same tissue of origin. Each cancer type—breast, lung melanoma, ovarian—falls into differing subtypes, and each subtype can be further divided; even within a subtype, each individual cancer is unique. This multiplicity of cancers has long been an obstacle to successful treatments.
But at their core, all cancers have this similarity: a gene in some region of the chromosome has mutated and is causing the cell to malfunction and divide uncontrollably. This basic understanding of cancer and the role of genes, long ago identified by biologists and geneticists, shows the complexity of cancer but also suggests a path forward. Find the gene causing the cancer, called a driver gene, then find a drug that works on that gene to inhibit its cancer-causing properties. (At least five different drugs target the Bcr-Abl mutation for example.)
Identifying a driver gene is not easy. Cancer cells are undergoing incredibly rapid mutation and evolution, and are compromised in regards to the body’s normally robust DNA repair mechanisms. Cancer cells have so much wrong with them that their entire DNA integrity and error correction is out of whack, resulting in the accumulation of multiple spurious mutations. Often entire sections of the chromosome are damaged; the difficulty is in zeroing in on the specific region likely to harbor a driver gene and then isolating the specific driver from surrounding mutated genes that don’t contribute to the cancer, the passenger genes.
Dana Pe’er, whose lab is working to apply computational methods to systems biology, likens hunting for a driver gene to investigating a plane crash. How is it possible, amidst the distributed wreckage, to find the one screw that failed?
New tools needed
For many years, tracking down the gene responsible was done in the lab using traditional bench biology tools to physically examine cells at the molecular level. Such methods are slow and lack the power and precision to identify weak and rare drivers. Biologists using traditional lab methods have identified only a limited of drivers.
As more cancer data accumulates, researchers are looking at computational methods. Classifying driver and passenger genes should be a classic machine-learning problem: Apply a supervised machine-learning approach to “learn” the features—attributes that are meaningful for a particular problem—that distinguish known driver genes and apply that method to identify other, still-unknown drivers. It’s a concept that rests on a solid statistical foundation and is being used successfully in other areas of everyday life to predict customer buying patterns and behavior, to detect fraud, and to recognize speech and handwriting.
The idea of applying machine-learning and other statistical methods to cancer research is not new. But the complexity of the biology relative to the abundance of data presents an obstacle. The traditional statistical methods at the heart of machine learning assume the number of samples is much larger than the number of features or parameters, an assumption that is not true in biology and cancer research. Tumor cohorts (a form of longitudinal study) typically number in the hundreds, while the number of potential drivers number in the tens of thousands. What is more, biology and cancer research also entail an extraordinarily large number of features; in the case of cancer there may be tens of thousands of features, many of which are expressions levels (genes can be expressed as either RNA or a protein product, with the level or amount of expression being highly informative). Tens of thousands of genes’ expressions are observed in a small number (hundreds to thousands) of samples.
Not all features are equal of course, and for each problem the set of meaningful features is different. One challenge in genomics is making the problem manageable by imposing constraints and honing in on those features—out of tens of thousands—that matter most. It’s a problem of feature selection, and for distinguishing driver from passenger genes, it requires both a strong understanding of machine learning methods and a deep appreciation of cancer biology and genomics.
A computer scientist learns biology
Felix Sanchez-Garcia began his graduate studies in 2008 in the Department of Computer Science, pursuing a master’s degree in machine learning. During his last semester, he began working in Dana Pe’er’s lab to provide statistical validation and support. At the time, he had little knowledge of biological sciences, but quickly got up to speed on the basics while working in the lab. After completing his master’s, he went on to pursue doctoral studies at Columbia, continuing to work in the Pe’er Lab while taking classes in molecular biology, cellular biology, cancer biology, and related topics.
As he gained a firm foundation of biological knowledge, he was in an excellent position to apply his machine learning expertise to biological problems, choosing for his PhD thesis the project of identifying additional drivers in breast cancer.
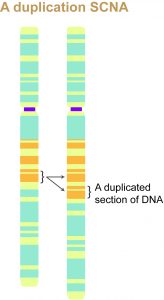
Breast cancers like many other cancers are the result of what is called a somatic copy number alteration (SCNA), which is a deletion or duplication of DNA within certain regions of the genome. SCNAs usually include tens of genes (as opposed to point mutations, which map to a single nucleotide pair in the DNA).
In an SCNA, DNA might be deleted or duplicated within certain regions of the chromosome. The marked region on the left is duplicated in the chromosome to the right. Such alterations to the DNA may or may not lead to cancer.
SCNAs are thought to recurrently affect close to 140 regions of the genome; almost every cancer genome will harbor a few. Not all SCNAs are harmful or significant. Many are simply random background alterations that have no ill effect.
For identifying significant SCNAs, there are existing algorithms but most lack the necessary resolution to identify driver regions small enough so it becomes possible to identify the single gene responsible for a cancer. Nor do SCNA-detection algorithms take into account that the alteration rate can differ greatly among different genomic regions; most (including GISTIC2) compute a null distribution across the entire genome to estimate the significance of alterations, meaning each region is independently evaluated against a global absolute.
Sanchez-Garcia suspected some regions might not score highly against the global absolute score but would nevertheless be significantly more altered than adjacent chromosomal regions. With help from Dylan Kotliar, Bo Juen Chen, and Uri David Akavia—all in the Pe’er Lab—he created a new algorithm called ISAR (Identification of Significantly Altered Regions) specifically to compare an alteration only with nearby, surrounding alterations. This measure of local alteration became an additional data point for identifying possible regions harboring driver genes.
Applying this methodology to 785 breast cancer tumor samples (taken from The Cancer Genome Atlas, or TCGA), ISAR identified 83 significant regions, more than double the 30 regions previously reported. Those regions found by ISAR included previously known regions as well as additional ones, including several containing known oncogenes (genes having the potential to cause cancer), thus providing strong evidence that the algorithm was making accurate predictions.
Looking to the data
With ISAR supplying a list of regions likely to harbor driver genes, Sanchez-Garcia and Pe’er then moved to the next step: identifying driver genes within those regions.
Standard classification approaches rely on an initial list of sample drivers and passengers to train the model. The problem here is that the list of known drivers is relatively small and strongly biased also (toward kinases and extreme phenotypes).
Sanchez-Garcia opted for a different approach, one that would look to the data itself to determine significant features. This more data-centric approach of course works best when data is plentiful, which is not the case for any single cancer data source. The solution was to incorporate different data sources, measuring different aspects of each cancer to obtain a range of different data types.
One data source was the primary tumor data, which is measured directly from patient tumors. From this data source Sanchez-Garcia obtained copy number, point mutations, and gene expression levels. A second data source was from cell lines, which is data originally derived from tumors but modified for lab study. For cell lines, he derived copy number, gene expression levels, and functional RNAi screening data, which provides functional information about the genome by knocking out each individual gene in a cell line.
Together this diverse set of data produced a single candidate driver score.
While previous methods had also integrated different data sources, particularly patient data and functional lab cell line, they tended to look for intersection between data sources, in effect narrowing the data by omitting non-overlapping data. Wanting to use more not less data, Sanchez-Garcia and Pe’er designed a new driver-detection algorithm called Helios with the aim of integrating the data sources in a more intelligent, complementary way.
It was here Sanchez-Garcia’s machine-learning expertise came most prominently into play. Using the technique of transfer learning—where information learned in one task is applied to another—Sanchez-Garcia initiates Helios with a small number of likely drivers, specifically those nearest the most frequently altered segment of an ISAR-identified region. Taking an unbiased approach, Helios learns the features of these strong drivers, and then looks for complementary patterns from the additional genetic and genomic data to discover additional features. If evidence for a feature in one data source is weak, it may be stronger in another. In this way, drivers with clearer signal help learn features to improve performance in cases with less obvious signal. Together the different features identify drivers that one feature by itself could not.
It’s an iterative approach where the algorithm is run multiple times. The first iteration picks up the strongest signals, and this information gets incorporated into the next iteration. Each iteration thus incorporates the new information the model learned in the previous iteration to pick up additional signal. Each subsequent iteration works off slightly more refined information than did the previous iteration. Helios keeps iterating through the data, producing drivers with a weaker but still distinct signal until there is no more signal to be found.
It is a statistically rigorous framework for combining multiple signals that might lack power individually. At the end, Helios produces a list of candidate drivers, ranked according to the probability each is a true cancer driver.
Shockingly accurate results
Validation was systematic and comprehensive. The researchers simply let the algorithm run to see what drivers it could discover. Of all predictions, researchers took the top scoring 17 for validation.
In seven of the 17 regions, the top Helios-identified gene was a bona-fide breast cancer oncogene (ERBB2, CCND1, ZNF217, Myc, miR-21, FGFR2, and IGF1R). Each scored well above the next best scoring gene. Myc’s Helios score, for example, was 100 times greater than the second best gene in the region. In the additional 10 regions, there was no known breast cancer driver so in each case researchers performed in vitro validation, finding that 10 of the 12 candidates selected in those 10 regions (in two regions, there were two equally scoring genes and both were selected for validation) provided a selective advantage for breast cancer cells.
Reflecting on these results, Dr. Pe’er explains, “This is the first and largest scale systematic validation undertaken for an algorithm of this type, and its accuracy is unprecedented. Its ability to reveal so many new cancer drivers is due to the fact that our hypotheses were generated in an unsupervised way using statistical criteria, rather than by cherry-picking our candidates based on prior biological knowledge. The experimental results show that Helios is a very robust algorithm that can generate biological insights that would be extremely difficult to produce in any other way.”
With ISAR selecting regions and Helios pinpointing the most likely driver within each region, Sanchez-Garcia and Pe’er were able to double the number of drivers implicated in breast cancer. The number of high-frequency drivers (seen in >5% patients, the ones that are more likely to be followed up with drug development), increased from 15 to 29.
The accurate results demonstrated by Helios hold out hope for more effective and personalized treatments for breast cancer and suggest which drugs should be developed. It is only the first step. The next step, already begun, will be to design therapies for patients.
The success of Helios demonstrates the validity of statistical modeling and other computational methods in discovering and treating disease through pure analysis of the data alone. Except for validating results in the lab, researchers worked only with data.
It is an approach not limited to breast cancer. In the same way Helios learns the weight of each feature directly from diverse breast-cancer data, it can do the same for other cancers. Already Sanchez-Garcia has been contacted by others interested in applying Helios to different malignancies to replicate the same success Helios has obtained in breast cancer: integrating diverse cancer data sets to identify and increase the number of cancer drivers.
Dana Pe’er is Associate Professor in the Department of Biological Sciences at Columbia University and regarded as a leading researcher in computational systems biology. Her research focuses on understanding the organization, function, and evolution of molecular networks, particularly how genetic variations alter the regulatory network and how these genetic variations can cause cancer.
Her lab at Columbia develops computational methods to integrate diverse high throughput data with a goal to personalize cancer care.
Pe’er received her bachelor’s degree in mathematics in 1995, her master’s degree in 1999, and her PhD in computer science in 2003, all from the Hebrew University of Jerusalem. Her PhD work used machine learning and Bayesian networks to automatically infer regulatory relations between genes, using genomics data.
In 2014 Pe’er won an ISCB Overton Prize Award. She is also the recipient of the Burroughs Wellcome Fund Career Award, NIH Directors New Innovator Award, NIH Directors Pioneer Award, NSF CAREER award, Stand Up To Cancer Innovative Research Grant, and a Packard Fellow in Science and Engineering.
During the last months leading up to completing his thesis he was living in Cambridge, England, and held the position of Visiting Scholar at the University of Cambridge.
His PhD now complete, Sanchez-Garcia, still in Cambridge, is taking time off from academia to work in an industry job in which he can apply his machine learning skills. Previously working as a data scientist at Expedia, he was recently named the chief data scientist at London’s Guardian newspaper.
He hasn’t left biology completely behind, however. He continues to collaborate with the Pe’er Lab, and is also contributing to research at the Sanger Institute. He remains particularly excited about an upcoming paper that will extend the work described in the Helios paper, remarking, “We’re in the middle of it and there’s still a lot of work to do, but I think if we can get it right, we will be able to identify new therapeutic approaches that could potentially have an enormous benefit to patients.”
The celebrated theorem of John Nash states that every game has an equilibrium point. During the past decade, computer theorists have investigated the computational complexity of finding exact or approximate Nash equilibria in various types of games. For anonymous games with a bounded number of strategies, an impressive sequence of papers by Constantinos Daskalakis and Christos Papadimitriou show that an approximate Nash equilibrium can be computed efficiently. (Anonymous games are an important class of multiplayer games where there are no special players; a single player’s payoff depends only on the player’s own strategy and the number—but not the identity—of other players playing each strategy. An example might be someone’s choice of attending a departmental meeting; if enough people attend, no matter who they may be, others may also decide to attend to avoid being conspicuously absent.)
Anthi Orfanou
However, the possibility of efficiently computing an exact Nash equilibrium (or an approximate one with an exponentially high accuracy) in such games remained open, though it was conjectured to be hard. In “On the Complexity of Nash Equilibria in Anonymous Games,” Columbia computer scientists Xi Chen, and Anthi Orfanou along with David Durfee (Georgia Institute of Technology) provide the proof that computing an exact equilibrium in anonymous games with a bounded number of strategies is indeed hard, and that the results obtained by Daskalakis and Papadimitriou cannot be extended to find a highly accurate Nash equilibrium.
While the paper’s result came as no surprise, it was not immediately obvious how to obtain such a proof, for which one needs to embed some other problem known to be hard. It’s not easy to embed things in anonymous games due to their highly symmetric payoff structure. Chen and his coauthors were however able to focus on a carefully picked class of anonymous games in which different players play a particular strategy with probabilities of different scales. This allowed them to analyze Nash equilibria of these games and embed things in them. This method may be applied when investigating the computational complexity of other types of games.
A natural framework for many types of data sets is to view them as a collection of (input,output) pairs. It is often important to know whether a data set is monotone or not, in the following sense: does increasing the input value always cause the output value to increase? Or can it sometimes cause the output to increase and sometimes to decrease? The standard way to check a data set for monotonicity is to scan through all the (input,output) pairs. However, this brute-force method is too inefficient for today’s massive high-dimensional data sets, when it is computationally too expensive to make even a single complete pass over the data.
In some situations, it suffices to know only whether a data set is monotone versus far from monotone. In this case, the key question becomes how many queries are needed to differentiate the two cases. The 1998 paper “Testing Monotonicity” (by Oded Goldreich, Shafi Goldwasser, Eric Lehman, and Dana Ron) gives an algorithm that requires only n queries for n-dimensional datasets (with 2n binary data entries), setting an upper bound on the query complexity of testing monotonicity of n-variable Boolean functions. Subsequent work by Deeparnab Chakrabarty and C. Seshadhri recently pushed the number of queries down from n to n7/8.
In the paper “Boolean function monotonicity testing requires (almost) n1/2 non-adaptive queries,” Xi Chen, Anindya De (IAS), Rocco A. Servedio, and Li-Yang Tan (UC Berkeley) studied lower bounds on the number of queries required for monotonicity testing. Their paper proves a lower bound of n1/2⁻c, for any positive constant c, on the query complexity of two-sided error non-adaptive algorithms for testing whether an n-variable Boolean function is monotone versus far from monotone. This bound of n1/2 is also conjectured to be tight. Their result represents an improvement over the previous lower bound of n1/5 by Chen, Servedio, and Tan in 2014.
Update: After this paper was announced, a new result by Subhash Khot, Dor Minzer, and Muli Safra improves the upper bound to n1/2 as conjectured, matching the lower bound of Chen, De, Servedio, and Tan.
Indistinguishability obfuscation is the process of encrypting software so it performs a function without the software itself being intelligible. Indistinguishability obfuscation was long thought to be impossible, but in 2013, six researchers (Sanjam Garg, Craig Gentry, Shai Halevi, Mariana Raykova, Amit Sahai, Brent Waters) published a paper that describes the first plausible candidate indistinguishability obfuscator using a new type of mathematical structure called multilinear maps. This candidate allows for obfuscation of any polynomially sized circuit.
While circuits represent a general model of computation, they have the drawback that the size of a circuit description of a computation is proportional to the running time of a computation. This might be much longer than the time required to simply describe the computation in a different form.
“Indistinguishability Obfuscation for Turing Machines with Unbounded Memory“, co-authored by Allison Bishop along with Venkata Koppula (University of Texas at Austin) and Brent Waters (University of Texas at Austin), replaces circuits (where the input length is fixed) with a Turing machine to show conceptually what can be done for varying input sizes. The proof of concept described in the paper shows how obfuscation time grows polynomially with the machine description size, |M|, as opposed to its worst case running time T. In addition to serving as its own application, such a construction paves the way for other applications such as delegation of computation.
Given two graphs with the same number of nodes and edges but arranged differently, how efficiently can an algorithm determine whether the two graphs are isomorphic? The problem looks innocent, and there are many different heuristics for finding features that might differentiate two graphs. In practice these heuristics run efficiently, but it cannot be proved they always give the right answer in polynomial time. This graph isomorphism problem is notorious for its unresolved complexity status, and there has been no improvement on the exp(~O(n1/2))-time algorithm obtained more than three decades ago.
In “In Faster Canonical Forms for Primitive Coherent Configurations,” Xiaorui Sun (Columbia University) and John Wilmes (University of Chicago), both PhD students, look at a special family of graphs, primitive coherent configurations (PCCs), which are edge-colored digraphs with certain regularity structures. Sun and Wilmes develop a new combinatorial structure theory for PCCs that in particular demonstrates the presence of “asymptotically uniform clique geometries” in PCCs with a certain range of parameters. Their results imply a faster algorithm for the isomorphism testing of PCCs with running time exp(~O(n1/3)) and the same upper bound on the number of automorphisms of PCCs, making the first progress in 33 years on a 1982 conjecture of Babai.
Changxi Zheng, assistant professor of computer science, is the recipient of a National Science Foundation (NSF) CAREER Award for his proposal to create realistic, computer-generated sounds for immersive virtual realities. The five-year, $500,000 grant will fund his proposal “Simulating Nonlinear Audiovisual Dynamics for Virtual Worlds and Interactive Applications.”
“I am honored by the award and excited about the research it supports,” says Zheng, who is co-directing the Columbia Computer Graphics Group (C2G2) in the Columbia Vision + Graphics Center. “The algorithms and tools used today for constructing immersive virtual realities are inherently appearance-oriented, modeling only geometry or visible motion. Sound is usually added manually and only as an afterthought. So there is a clear gap.”
While computer-generated imagery has made tremendous progress in recent years to attain high levels of realism, the same efforts have not yet been applied to computer-generated sound. Zheng is among the first working in the area of dynamic, physics-based computational sound for immersive environments, and his proposal will look to tightly integrate the visual and audio components of simulated environments. It will represent a change from today’s practices where digital sound is usually created and recorded separately of the action and then dropped in when appropriate. In Zheng’s proposal, computational sound will be automatically generated using physics-based simulation methods and fully synchronized with the associated motion.
“What I propose to do about sound synthesis is analogous to the existing image rendering methods for creating photorealistic images,” says Zheng. “We would like to create audiovisual animations and virtual environments using computational methods.”
“In addition to the realism, sound that is not tightly coupled with motion loses some of its impact and detracts from the overall immersive experience,” says Zheng. “Attaining any type of realism in virtual worlds requires synchronizing the audio and visual components. What the user hears should spring directly and naturally from what the user sees.”
It will take new mathematical models and new computational methods. Sound is a physical phenomenon, and creating sound by computer requires understanding and simulating all the motions and forces that go into producing sound. Computational methods will have to replicate everything from the surface vibrations on an object that produce the pressure waves we hear as sound, while taking into account how frequency, pitch, and volume are affected by the object’s size, shape, weight, surface textures, and countless other variables. The way sound propagates differs also, depending on whether sound waves travel through air or water and what obstructions, or other sound waves, get in the way. It’s a dynamic situation where a slight adjustment to one variable produces nonlinear changes in another.
Zheng’s system will have to tackle not only the physical phenomena but do so in a computationally efficient way so there is no delay between an action and the resulting sound. “Realistic virtual environments require a great many complex computations. We will need fast, cost-effective algorithms. We need also to identify what calculations can be approximated while still maintaining realism.”
The computational sound is just the beginning. Zheng foresees his proposal as laying the groundwork for building interactive applications and spurring new techniques for efficient digital production and engineering design. Education outreach for disseminating the information learned from the research is also part of the proposal, with workshops planned for high-school students to encourage their interest in STEM fields, and with college courses for teaching undergraduate and graduate researchers how to build audiovisual computational technologies.
For now, numerous challenges exist toward computationally synthesizing realistic sound but if the research is successful, it will enable new virtual-world applications with fully synchronized audiovisual effects, lead to new ways of creating and editing multimedia content, and bring computer-generated sound into the future.
BS, Shanghai Jiaotong University (China), 2005; PhD, Cornell University, 2012
Dean Boyce's statement on amicus brief filed by President Bollinger
President Bollinger announced that Columbia University along with many other academic institutions (sixteen, including all Ivy League universities) filed an amicus brief in the U.S. District Court for the Eastern District of New York challenging the Executive Order regarding immigrants from seven designated countries and refugees. Among other things, the brief asserts that “safety and security concerns can be addressed in a manner that is consistent with the values America has always stood for, including the free flow of ideas and people across borders and the welcoming of immigrants to our universities.”
This recent action provides a moment for us to collectively reflect on our community within Columbia Engineering and the importance of our commitment to maintaining an open and welcoming community for all students, faculty, researchers and administrative staff. As a School of Engineering and Applied Science, we are fortunate to attract students and faculty from diverse backgrounds, from across the country, and from around the world. It is a great benefit to be able to gather engineers and scientists of so many different perspectives and talents – all with a commitment to learning, a focus on pushing the frontiers of knowledge and discovery, and with a passion for translating our work to impact humanity.
I am proud of our community, and wish to take this opportunity to reinforce our collective commitment to maintaining an open and collegial environment. We are fortunate to have the privilege to learn from one another, and to study, work, and live together in such a dynamic and vibrant place as Columbia.
Sincerely,
Mary C. Boyce
Dean of Engineering
Morris A. and Alma Schapiro Professor

 Henning Schulzrinne, the Julian Clarence Levi Professor of Mathematical Methods and Computer Science, has been named a 2014 ACM (Association for Computing Machinery) Fellow in recognition of “contributions to the design of protocols, applications, and algorithms for Internet multimedia.” He is one of 47 to be so recognized in 2014 for contributions to computing.
Henning Schulzrinne, the Julian Clarence Levi Professor of Mathematical Methods and Computer Science, has been named a 2014 ACM (Association for Computing Machinery) Fellow in recognition of “contributions to the design of protocols, applications, and algorithms for Internet multimedia.” He is one of 47 to be so recognized in 2014 for contributions to computing.