|
Machine learning applied to cancer: A PhD student doubles the number of breast cancer drivers
Identifying the specific genes that cause a cancer—the so-called driver genes—is a central goal in cancer research. The more drivers identified, the more options to treat the cancer with a drug known to act on that gene. But identifying a specific driver is not easy, especially when entire sections of the chromosome are damaged. Traditional biological tools, slow and imprecise, have identified only a limited number. As cancer "big data" accumulates, researchers are turning to computational methods, specifically machine learning, to identify additional drivers. But it's not immediately clear how to apply these methods to genetic data. Machine learning expects a lot of data, but biological data is relatively sparse compared to other application domains in machine learning. In a paper published last November in Cell, Felix Sanchez-Garcia, a computer science PhD student, describes a new algorithm that integrates data from multiple diverse sources, using the strong features in one to bolster weaker evidence in the other. Applied to breast cancer patients, the algorithm doubled the number of identified driver genes.
Cancer is a heterogeneous disease, and no two cancers are the same even when they occur at the same tissue of origin. Each cancer type—breast, lung melanoma, ovarian—falls into differing subtypes, and each subtype can be further divided; even within a subtype, each individual cancer is unique. This multiplicity of cancers has long been an obstacle to successful treatments.
But at their core, all cancers have this similarity: a gene in some region of the chromosome has mutated and is causing the cell to malfunction and divide uncontrollably. This basic understanding of cancer and the role of genes, long ago identified by biologists and geneticists, shows the complexity of cancer but also suggests a path forward. Find the gene causing the cancer, called a driver gene, then find a drug that works on that gene to inhibit its cancer-causing properties. (At least five different drugs target the Bcr-Abl mutation for example.)
Identifying a driver gene is not easy. Cancer cells are undergoing incredibly rapid mutation and evolution, and are compromised in regards to the body's normally robust DNA repair mechanisms. Cancer cells have so much wrong with them that their entire DNA integrity and error correction is out of whack, resulting in the accumulation of multiple spurious mutations. Often entire sections of the chromosome are damaged; the difficulty is in zeroing in on the specific region likely to harbor a driver gene and then isolating the specific driver from surrounding mutated genes that don't contribute to the cancer, the passenger genes.
Dana Pe'er, whose lab is working to apply computational methods to systems biology, likens hunting for a driver gene to investigating a plane crash. How is it possible, amidst the distributed wreckage, to find the one screw that failed?
New tools needed
For many years, tracking down the gene responsible was done in the lab using traditional bench biology tools to physically examine cells at the molecular level. Such methods are slow and lack the power and precision to identify weak and rare drivers. Biologists using traditional lab methods have identified only a limited of drivers.
As more cancer data accumulates, researchers are looking at computational methods. Classifying driver and passenger genes should be a classic machine-learning problem: Apply a supervised machine-learning approach to "learn" the features—attributes that are meaningful for a particular problem—that distinguish known driver genes and apply that method to identify other, still-unknown drivers. It's a concept that rests on a solid statistical foundation and is being used successfully in other areas of everyday life to predict customer buying patterns and behavior, to detect fraud, and to recognize speech and handwriting.
The idea of applying machine-learning and other statistical methods to cancer research is not new. But the complexity of the biology relative to the abundance of data presents an obstacle. The traditional statistical methods at the heart of machine learning assume the number of samples is much larger than the number of features or parameters, an assumption that is not true in biology and cancer research. Tumor cohorts (a form of longitudinal study) typically number in the hundreds, while the number of potential drivers number in the tens of thousands. What is more, biology and cancer research also entail an extraordinarily large number of features; in the case of cancer there may be tens of thousands of features, many of which are expressions levels (genes can be expressed as either RNA or a protein product, with the level or amount of expression being highly informative). Tens of thousands of genes' expressions are observed in a small number (hundreds to thousands) of samples.
Not all features are equal of course, and for each problem the set of meaningful features is different. One challenge in genomics is making the problem manageable by imposing constraints and honing in on those features—out of tens of thousands—that matter most. It's a problem of feature selection, and for distinguishing driver from passenger genes, it requires both a strong understanding of machine learning methods and a deep appreciation of cancer biology and genomics.
A computer scientist learns biology
Felix Sanchez-Garcia began his graduate studies in 2008 in the Department of Computer Science, pursuing a master's degree in machine learning. During his last semester, he began working in Dana Pe'er's lab to provide statistical validation and support. At the time, he had little knowledge of biological sciences, but quickly got up to speed on the basics while working in the lab. After completing his master's, he went on to pursue doctoral studies at Columbia, continuing to work in the Pe'er Lab while taking classes in molecular biology, cellular biology, cancer biology, and related topics.
As he gained a firm foundation of biological knowledge, he was in an excellent position to apply his machine learning expertise to biological problems, choosing for his PhD thesis the project of identifying additional drivers in breast cancer.
Breast cancers like many other cancers are the result of what is called a somatic copy number alteration (SCNA), which is a deletion or duplication of DNA within certain regions of the genome. SCNAs usually include tens of genes (as opposed to point mutations, which map to a single nucleotide pair in the DNA).
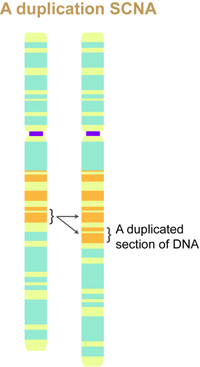
In an SCNA, DNA might be deleted or duplicated within certain regions of the chromosome. The marked region on the left is duplicated in the chromosome to the right. Such alterations to the DNA may or may not lead to cancer.
SCNAs are thought to recurrently affect close to 140 regions of the genome; almost every cancer genome will harbor a few. Not all SCNAs are harmful or significant. Many are simply random background alterations that have no ill effect.
For identifying significant SCNAs, there are existing algorithms but most lack the necessary resolution to identify driver regions small enough so it becomes possible to identify the single gene responsible for a cancer. Nor do SCNA-detection algorithms take into account that the alteration rate can differ greatly among different genomic regions; most (including GISTIC2) compute a null distribution across the entire genome to estimate the significance of alterations, meaning each region is independently evaluated against a global absolute.
Sanchez-Garcia suspected some regions might not score highly against the global absolute score but would nevertheless be significantly more altered than adjacent chromosomal regions. With help from Dylan Kotliar, Bo Juen Chen, and Uri David Akavia—all in the Pe'er Lab—he created a new algorithm called ISAR (Identification of Significantly Altered Regions) specifically to compare an alteration only with nearby, surrounding alterations. This measure of local alteration became an additional data point for identifying possible regions harboring driver genes.
Applying this methodology to 785 breast cancer tumor samples (taken from The Cancer Genome Atlas, or TCGA), ISAR identified 83 significant regions, more than double the 30 regions previously reported. Those regions found by ISAR included previously known regions as well as additional ones, including several containing known oncogenes (genes having the potential to cause cancer), thus providing strong evidence that the algorithm was making accurate predictions.
Looking to the data
With ISAR supplying a list of regions likely to harbor driver genes, Sanchez-Garcia and Pe'er then moved to the next step: identifying driver genes within those regions.
Standard classification approaches rely on an initial list of sample drivers and passengers to train the model. The problem here is that the list of known drivers is relatively small and strongly biased also (toward kinases and extreme phenotypes).
Sanchez-Garcia opted for a different approach, one that would look to the data itself to determine significant features. This more data-centric approach of course works best when data is plentiful, which is not the case for any single cancer data source. The solution was to incorporate different data sources, measuring different aspects of each cancer to obtain a range of different data types.
One data source was the primary tumor data, which is measured directly from patient tumors. From this data source Sanchez-Garcia obtained copy number, point mutations, and gene expression levels. A second data source was from cell lines, which is data originally derived from tumors but modified for lab study. For cell lines, he derived copy number, gene expression levels, and functional RNAi screening data, which provides functional information about the genome by knocking out each individual gene in a cell line.
Together this diverse set of data produced a single candidate driver score.
While previous methods had also integrated different data sources, particularly patient data and functional lab cell line, they tended to look for intersection between data sources, in effect narrowing the data by omitting non-overlapping data. Wanting to use more not less data, Sanchez-Garcia and Pe'er designed a new driver-detection algorithm called Helios with the aim of integrating the data sources in a more intelligent, complementary way.
It was here Sanchez-Garcia's machine-learning expertise came most prominently into play. Using the technique of transfer learning—where information learned in one task is applied to another—Sanchez-Garcia initiates Helios with a small number of likely drivers, specifically those nearest the most frequently altered segment of an ISAR-identified region. Taking an unbiased approach, Helios learns the features of these strong drivers, and then looks for complementary patterns from the additional genetic and genomic data to discover additional features. If evidence for a feature in one data source is weak, it may be stronger in another. In this way, drivers with clearer signal help learn features to improve performance in cases with less obvious signal. Together the different features identify drivers that one feature by itself could not.
It's an iterative approach where the algorithm is run multiple times. The first iteration picks up the strongest signals, and this information gets incorporated into the next iteration. Each iteration thus incorporates the new information the model learned in the previous iteration to pick up additional signal. Each subsequent iteration works off slightly more refined information than did the previous iteration. Helios keeps iterating through the data, producing drivers with a weaker but still distinct signal until there is no more signal to be found.
It is a statistically rigorous framework for combining multiple signals that might lack power individually. At the end, Helios produces a list of candidate drivers, ranked according to the probability each is a true cancer driver.
Shockingly accurate results
Validation was systematic and comprehensive. The researchers simply let the algorithm run to see what drivers it could discover. Of all predictions, researchers took the top scoring 17 for validation.
In seven of the 17 regions, the top Helios-identified gene was a bona-fide breast cancer oncogene (ERBB2, CCND1, ZNF217, Myc, miR-21, FGFR2, and IGF1R). Each scored well above the next best scoring gene. Myc's Helios score, for example, was 100 times greater than the second best gene in the region. In the additional 10 regions, there was no known breast cancer driver so in each case researchers performed in vitro validation, finding that 10 of the 12 candidates selected in those 10 regions (in two regions, there were two equally scoring genes and both were selected for validation) provided a selective advantage for breast cancer cells.
Reflecting on these results, Dr. Pe'er explains, "This is the first and largest scale systematic validation undertaken for an algorithm of this type, and its accuracy is unprecedented. Its ability to reveal so many new cancer drivers is due to the fact that our hypotheses were generated in an unsupervised way using statistical criteria, rather than by cherry-picking our candidates based on prior biological knowledge. The experimental results show that Helios is a very robust algorithm that can generate biological insights that would be extremely difficult to produce in any other way."
With ISAR selecting regions and Helios pinpointing the most likely driver within each region, Sanchez-Garcia and Pe'er were able to double the number of drivers implicated in breast cancer. The number of high-frequency drivers (seen in >5% patients, the ones that are more likely to be followed up with drug development), increased from 15 to 29.
The accurate results demonstrated by Helios hold out hope for more effective and personalized treatments for breast cancer and suggest which drugs should be developed. It is only the first step. The next step, already begun, will be to design therapies for patients.
The success of Helios demonstrates the validity of statistical modeling and other computational methods in discovering and treating disease through pure analysis of the data alone. Except for validating results in the lab, researchers worked only with data.
It is an approach not limited to breast cancer. In the same way Helios learns the weight of each feature directly from diverse breast-cancer data, it can do the same for other cancers. Already Sanchez-Garcia has been contacted by others interested in applying Helios to different malignancies to replicate the same success Helios has obtained in breast cancer: integrating diverse cancer data sets to identify and increase the number of cancer drivers.
For more information about Helios and ISAR, see "Integration of Genomic Data Enables Selective Discovery of Breast Cancer Drivers" (in Cell, November 2014).
-Linda Crane
Posted 6/25/2015
|
About the researchers

Dana Pe'er is Associate Professor in the Department of Biological Sciences at Columbia University and regarded as a leading researcher in computational systems biology. Her research focuses on understanding the organization, function, and evolution of molecular networks, particularly how genetic variations alter the regulatory network and how these genetic variations can cause cancer.
Her lab at Columbia develops computational methods to integrate diverse high throughput data with a goal to personalize cancer care.
Pe'er received her bachelor's degree in mathematics in 1995, her master's degree in 1999, and her PhD in computer science in 2003, all from the Hebrew University of Jerusalem. Her PhD work used machine learning and Bayesian networks to automatically infer regulatory relations between genes, using genomics data.
In 2014 Pe'er won an ISCB Overton Prize Award. She is also the recipient of the Burroughs Wellcome Fund Career Award, NIH Directors New Innovator Award, NIH Directors Pioneer Award, NSF CAREER award, Stand Up To Cancer Innovative Research Grant, and a Packard Fellow in Science and Engineering.

Felix Sanchez-Garcia received his PhD in Computer Science from Columbia Engineering in July 2014 for the research summarized in this article. For the full thesis, see "Integration of Genomic Data Enables Selective Discovery of Breast Cancer Drivers," which was published in Cell, November 2014).
During the last months leading up to completing his thesis he was living in Cambridge, England, and held the position of Visiting Scholar at the University of Cambridge.
His PhD now complete, Sanchez-Garcia, still in Cambridge, is taking time off from academia to work in an industry job in which he can apply his machine learning skills. Previously working as a data scientist at Expedia, he was recently named the chief data scientist at London's Guardian newspaper.
He hasn't left biology completely behind, however. He continues to collaborate with the Pe'er Lab, and is also contributing to research at the Sanger Institute. He remains particularly excited about an upcoming paper that will extend the work described in the Helios paper, remarking, "We're in the middle of it and there's still a lot of work to do, but I think if we can get it right, we will be able to identify new therapeutic approaches that could potentially have an enormous benefit to patients."
|